

Was ist PKU (Phenylketonurie)?
Symptome und Diagnose
Die Phenylketonurie, kurz PKU, ist eine seltene, autosomal-rezessiv vererbte Störung des Phenylalanin (Phe)-Stoffwechsels, mit der etwa 1 von 15.000 Menschen auf der Welt geboren wird.
Von PKU Betroffene können eine Aminosäure namens Phenylalanin (Phe), nicht abbauen, die sich folglich im Körper anreichert, toxisch ist und schwere Hirnschäden verursachen kann.
Die Behandlung muss lebenslang durchgeführt werden und besteht in einer Phe-armen Diät, d. h. einer eiweißarmen Diät, die mit Phe-freien Aminosäurenmischungen ergänzt wird, um ein normales Wachstum und eine gute Entwicklung zu gewährleisten.
Eine frühzeitige Diagnose und eine sofortige Therapieeinleitung ermöglichen es PKU-Betroffenen, gesund aufzuwachsen, den erwarteten Bildungsstandard zu erreichen und als Erwachsene ein unabhängiges Leben zu führen.1
Ursachen von Phenylketonurie und Klassifizierung
Phenylketonurie wird durch Varianten des Gens, das die Phenylalaninhydroxylase (PAH) codiert, verursacht. PAH ist ein Leberenzym, das den Cofaktor Tetrahydrobiopterin (BH4) benötigt, um Phenylalanin (Phe) in Tyrosin (Tyr) umzuwandeln. Ein Mangel an PAH oder seinem Cofaktor BH4 führt zu erhöhten Phe-Konzentrationen im Blut und toxischen Konzentrationen im Gehirn.
Wenn sie nicht sorgfältig kontrolliert werden, können die erhöhten Phe-Werte im Blut zu Verhaltensstörungen und neurologischen Beeinträchtigungen führen.1
Wenn sie nicht sorgfältig kontrolliert werden, können die erhöhten Phe-Werte im Blut zu Verhaltensstörungen und neurologischen Beeinträchtigungen führen.1
Je nach Enzymdefekt und Schweregrad der Erkrankung sind verschiedene Formen der PKU mit unterschiedlichen klinischen Erscheinungsbildern beschrieben worden. Daher wurden verschiedene Klassifizierungen für PKU-Phänotypen erstellt. PKU kann in eine klassische PKU und mildere Formen der PKU (z. B. moderate PKU und milde PKU) unterteilt werden.
Die Definition des PKU-Phänotyps kann für die Festlegung von Behandlungsoptionen, z. B. für neue therapeutische Strategien, für die Beratung und die Prognose von Behandlungsergebnissen sowie für die Schwangerschaft von wesentlicher Bedeutung sein.2
PKU-Test: Wie wird Phenylketonurie diagnostiziert?
Der Test auf PKU wird in der Regel im Rahmen des Neugeborenenscreenings (NBS) etwa 24-72 Stunden nach der Geburt im Krankenhaus durchgeführt, wobei eine Blutprobe aus der Ferse entnommen und der Phe-Spiegel im Blut mit einem einfachen Labortest gemessen wird. Die Screeningprogramme für Neugeborene können sich von Land zu Land unterscheiden.
Was sind PKU-Symptome?
Neugeborene mit PKU haben zunächst keine Symptome. Bleibt die Erkrankung jedoch unbehandelt, wird die Entwicklung des Gehirns stark beeinträchtigt, und es kommt zu erheblichen geistigen Behinderungen und Verhaltensauffälligkeiten.1
Eine diätetische Behandlung führt bei PKU-Patienten zu guten kognitiven und psychiatrischen Ergebnissen, insbesondere wenn sie über die gesamte neurologische Entwicklung hinweg und bis weit ins Erwachsenenalter beibehalten wird.3
Es kann jedoch schwierig sein, die PKU-Diät ein Leben lang einzuhalten. Während die Einhaltung der Diät (Therapietreue) in der Kindheit oft sehr gut ist, nimmt sie im Laufe des Erwachsenwerdens immer mehr ab4. Dies ist wahrscheinlich auf die zunehmende Unabhängigkeit der Patienten von ihren Familien sowie auf psychologische und soziale Belastungen zurückzuführen.
Es kann jedoch schwierig sein, die PKU-Diät ein Leben lang einzuhalten. Während die Einhaltung der Diät (Therapietreue) in der Kindheit oft sehr gut ist, nimmt sie im Laufe des Erwachsenwerdens immer mehr ab4. Dies ist wahrscheinlich auf die zunehmende Unabhängigkeit der Patienten von ihren Familien sowie auf psychologische und soziale Belastungen zurückzuführen.
Bei vielen PKU-Betroffenen, die bereits im Säuglingsalter mit einer Ernährungsbehandlung begonnen haben (Frühbehandelte) und diese später absetzen oder ihre diätetische Kontrolle lockern, können Symptome wie Angst, Depression und kognitive Störungen auftreten.5
Kann man PKU vorbeugen?
PKU ist genetisch bedingt und kann nicht verhindert oder vermieden werden. Sie wird rezessiv von den Eltern auf das Kind vererbt, d. h. beide Elternteile müssen ein mutiertes PAH-Gen besitzen, um die Krankheit zu vererben. Diese Personen werden als Träger bezeichnet.
Wenn nur ein Elternteil das mutierte Gen trägt, wird das Kind keine PKU entwickeln.
Selbst wenn beide Elternteile das mutierte Gen tragen, kann es sein, dass ihr Kind keine PKU entwickelt. Allerdings besteht auch in diesem Fall eine 25%ige Wahrscheinlichkeit, dass beide Elternteile das mutierte Gen weitergeben und das Kind PKU bekommt.
Selbst wenn beide Elternteile das mutierte Gen tragen, kann es sein, dass ihr Kind keine PKU entwickelt. Allerdings besteht auch in diesem Fall eine 25%ige Wahrscheinlichkeit, dass beide Elternteile das mutierte Gen weitergeben und das Kind PKU bekommt.
PKU und Schwangerschaft
Schwangerschaften sollten bei PKU geplant werden. Frauen mit PKU können gesunde Kinder zur Welt bringen, solange sie sich während der Schwangerschaft strikt an ihre Phe-arme Diät halten.
Eine während der Schwangerschaft schlecht kontrollierte PKU und hohe Phenylalaninwerte können das ungeborene Kind schädigen und Geburtsfehler verursachen.
Eine während der Schwangerschaft schlecht kontrollierte PKU und hohe Phenylalaninwerte können das ungeborene Kind schädigen und Geburtsfehler verursachen.
Je näher der Phe-Spiegel im Blutplasma der Mutter am Normalwert liegt, desto unwahrscheinlicher sind diese Komplikationen. Aus diesem Grund wird empfohlen, einen Empfängnisversuch zu unternehmen, sobald der Phe-Spiegel im Zielbereich für eine Schwangerschaft liegt. Wenn dies nicht möglich ist, z. B. bei unerwarteten und ungeplanten Schwangerschaften, sollte eine sofortige Überweisung an das Stoffwechselzentrum erfolgen, insbesondere wenn die Phe-Werte nicht im Zielbereich liegen.
Darüber hinaus muss die Schwangerschaft sehr sorgfältig überwacht werden, auch wenn die Phe-Werte von Anfang an unter Kontrolle sind. Es wird mindestens eine Untersuchung pro Vierteljahr empfohlen, aber viele Kliniker sprechen sich für häufigere Untersuchungen aus, wobei die Intensität der Überwachung von den individuellen Bedürfnissen und der Stoffwechselkontrolle abhängig ist1
PKU-Behandlung
Da Phe als Aminosäure ein Baustein von Proteinen (Eiweißen) ist und in allen Proteinen und den meisten Nahrungsmitteln vorkommt, beinhaltet die Behandlung eine besonders eingeschränkte und lebenslange Diät, deren Ziel es ist, den Phe-Spiegel im Blut niedrig zu halten1,6. Ernährung bei PKU:

Grundsätzlich sind Fleisch, Fisch, Eier und Milchprodukte nicht erlaubt

Nudeln, Brot und Kekse sollten eiweißarme Spezialzubereitungen sein

Das meiste Gemüse und Obst muss gewogen werden
Die Hauptproteinquelle sind spezielle Proteinersatzprodukte, die in der Regel auf phenylalaninfreien Aminosäuren basieren und für die Förderung eines normalen Wachstums, die Vorbeugung von Eiweißmangel, die Bereitstellung einer Tyrosinquelle und die Optimierung der Kontrolle des Phenylalanins im Blut unerlässlich sind.6

Unerfüllte Bedürfnisse bei herkömmlichem Proteinersatz
Herkömmliche Eiweißersatzprodukte enthalten eine Mischung freier Aminosäuren und sollten entsprechend den europäischen Leitlinien in kleinen, häufigen Dosen mindestens drei- bis viermal gleichmäßig über den Tag verteilt eingenommen werden.1
Heutzutage gibt es eine große Auswahl an verschiedenen Geschmacksrichtungen und Darreichungsformen. Internationale PKU-Experten weisen jedoch darauf hin, dass noch Verbesserungen möglich sind, um PKU-Patienten zu unterstützen und einige unerfüllte Bedürfnisse zu befriedigen, die in Bezug auf Therapietreue und Stoffwechselkontrolle heute noch bestehen.7
Heutzutage gibt es eine große Auswahl an verschiedenen Geschmacksrichtungen und Darreichungsformen. Internationale PKU-Experten weisen jedoch darauf hin, dass noch Verbesserungen möglich sind, um PKU-Patienten zu unterstützen und einige unerfüllte Bedürfnisse zu befriedigen, die in Bezug auf Therapietreue und Stoffwechselkontrolle heute noch bestehen.7
1. Therapietreue
Das Aufwachsen mit Phenylketonurie kann manchmal schwierig sein und zu einer nicht sichtbaren Belastung für die Betroffenen werden.
Die tägliche Einhaltung der Diät kann das soziale Leben von PKU-Patienten bei den Mahlzeiten beeinträchtigen, und die regelmäßige Einnahme von Proteinersatzstoffen kann aufgrund ihrer Darreichungsform und Eigenschaften sehr schwierig sein.
Die Therapietreue wird auch durch die unangenehmen organoleptischen Eigenschaften von Aminosäuren (Geschmack, Geruch und anhaltender Nachgeschmack im Mund nach dem Verzehr)7 erschwert, wodurch das soziale Leben der Patienten stark beeinträchtigt wird.
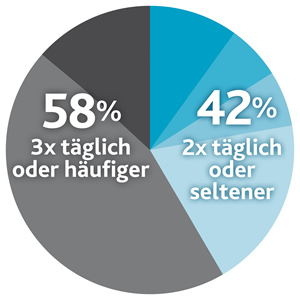 Darüber hinaus hat eine kürzlich durchgeführte Studie an insgesamt therapietreuen Patienten gezeigt, dass die Häufigkeit der Einnahme von Aminosäuremischungen bei der PKU-Behandlung eine heikle Rolle spielen kann, da 42 % der erwachsenen PKU-Patienten ihren gesamten Proteinersatz auf ≤2 Dosen/Tag aufteilen (im Gegensatz zu den in den europäischen Leitlinien empfohlenen 3 bis 4 Dosen).8
Darüber hinaus hat eine kürzlich durchgeführte Studie an insgesamt therapietreuen Patienten gezeigt, dass die Häufigkeit der Einnahme von Aminosäuremischungen bei der PKU-Behandlung eine heikle Rolle spielen kann, da 42 % der erwachsenen PKU-Patienten ihren gesamten Proteinersatz auf ≤2 Dosen/Tag aufteilen (im Gegensatz zu den in den europäischen Leitlinien empfohlenen 3 bis 4 Dosen).8
2. Stoffwechselkontrolle
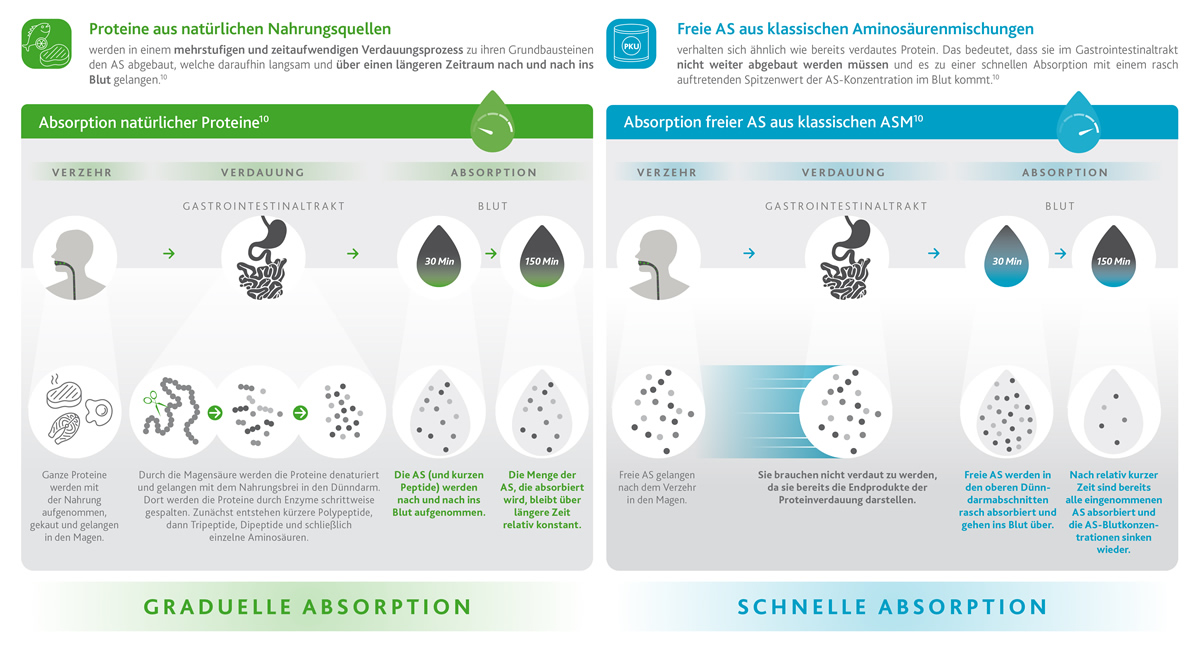
Ein weiterer wichtiger Aspekt, der bei der PKU-Behandlung zu berücksichtigen ist, betrifft die Art und Weise, wie Aminosäuren nach ihrer Aufnahme über Proteinersatzstoffe vom Körper absorbiert werden.
Personen ohne PKU nehmen natürliche Proteine mit der Nahrung auf (z. B. Fleisch, Fisch, Milchprodukte, Eier usw.), die im Laufe der Zeit verdaut und absorbiert werden (grüner Kasten), so dass der Aminosäurespiegel im Blut bis zu 150 Minuten (2,5 Stunden) konstant bleibt.
Bei PKU-Betroffenen müssen eiweißhaltige Lebensmittel durch freie Aminosäuren ersetzt werden, die in herkömmlichen Proteinersatzprodukten enthalten sind. Proteine und Aminosäuren verhalten sich jedoch sehr unterschiedlich!
Freie Aminosäuren sind wie bereits verdaute Proteine, so dass sie direkt für die Aufnahme durch den Darm verfügbar sind. Dies bedeutet, dass der Aminosäurespiegel im Blut unmittelbar nach dem Verzehr von Proteinersatzprodukten hoch ist9 und innerhalb von nur 30 Minuten einen Höchstwert erreicht, der bald darauf drastisch abfällt (blauer Kasten).1,10
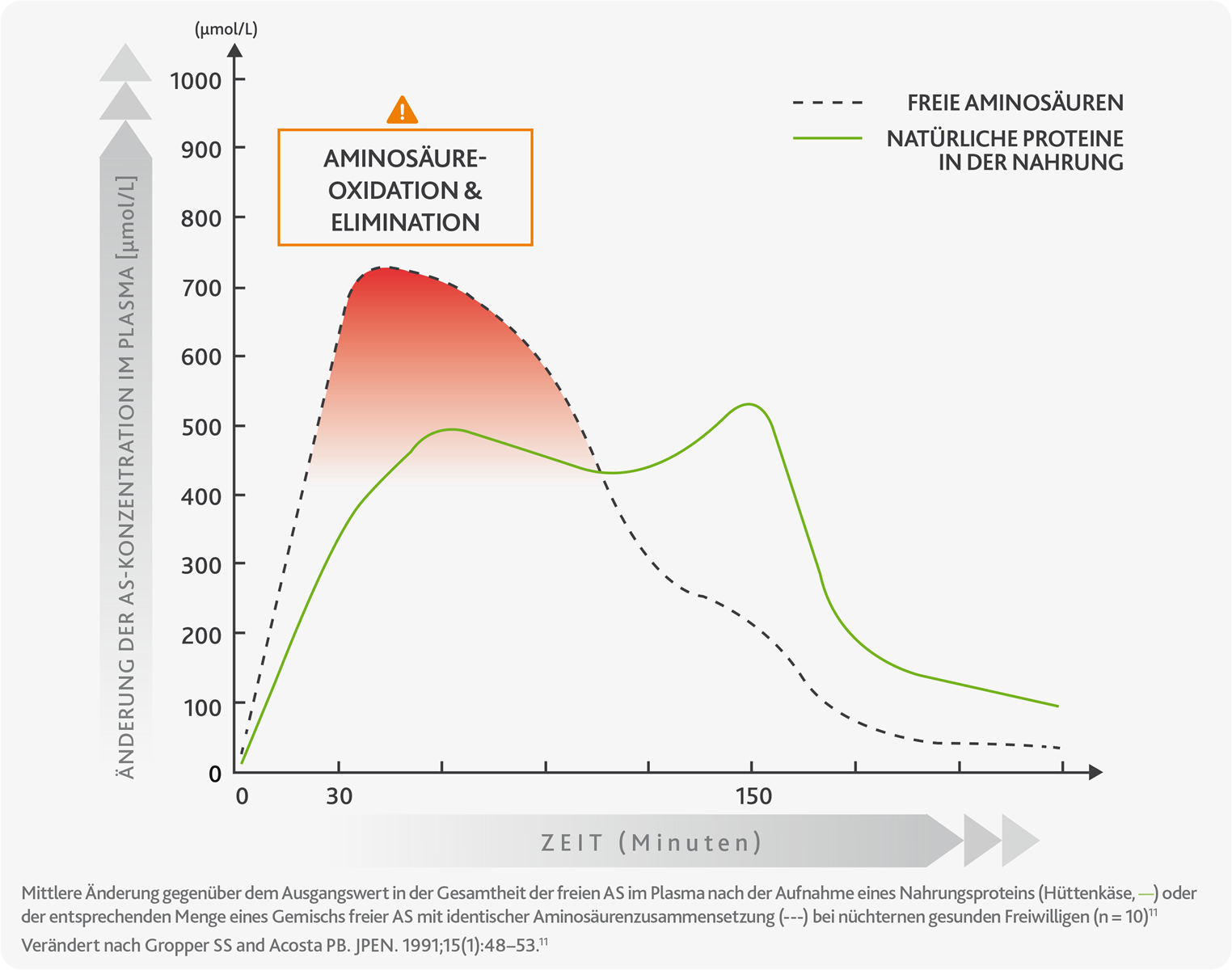
 Die Grafik zeigt detaillierter den Unterschied in der Absorptionskinetik zwischen natürlichem Protein (grüne Linie) und freien Aminosäuren (gestrichelte Linie), wobei deren hohe Ausscheidung kurz nach der Aufnahme (Oxidation, roter Bereich) und in den so genannten „Fastenperioden“, angezeigt wird, d. h. in den Zeitabschnitten, in denen die Menge an Aminosäuren im Körper sehr gering ist.10,11
Die Oxidation und schnelle Ausscheidung von Aminosäuren verhindert, dass der Körper sie effizient für physiologische Prozesse wie den Aufbau neuer Proteine nutzen kann, was sich auf den Stoffwechsel des Körpers auswirkt und folglich die Gesundheit beeinträchtigt.10,11
Die Grafik zeigt detaillierter den Unterschied in der Absorptionskinetik zwischen natürlichem Protein (grüne Linie) und freien Aminosäuren (gestrichelte Linie), wobei deren hohe Ausscheidung kurz nach der Aufnahme (Oxidation, roter Bereich) und in den so genannten „Fastenperioden“, angezeigt wird, d. h. in den Zeitabschnitten, in denen die Menge an Aminosäuren im Körper sehr gering ist.10,11
Die Oxidation und schnelle Ausscheidung von Aminosäuren verhindert, dass der Körper sie effizient für physiologische Prozesse wie den Aufbau neuer Proteine nutzen kann, was sich auf den Stoffwechsel des Körpers auswirkt und folglich die Gesundheit beeinträchtigt.10,11
 Die unzureichende Absorption von Aminosäuren und die Unfähigkeit des Körpers, sie effizient zu nutzen, ist der Grund, warum in den europäischen Leitlinien für PKU empfohlen wird, herkömmliche Proteinersatzstoffe in kleinen und häufigen Dosen mindestens 3- bis 4-mal täglich zu verzehren.
Die Gesamtproteinzufuhr sollte 40 % mehr als die von FAO/WHO/UNU festgelegten sicheren Eiweißmengen liefern1,6, um den Aminosäurespiegel im Blut konstant zu halten.
Die unzureichende Absorption von Aminosäuren und die Unfähigkeit des Körpers, sie effizient zu nutzen, ist der Grund, warum in den europäischen Leitlinien für PKU empfohlen wird, herkömmliche Proteinersatzstoffe in kleinen und häufigen Dosen mindestens 3- bis 4-mal täglich zu verzehren.
Die Gesamtproteinzufuhr sollte 40 % mehr als die von FAO/WHO/UNU festgelegten sicheren Eiweißmengen liefern1,6, um den Aminosäurespiegel im Blut konstant zu halten.
Dies ist jedoch oft sehr schwer zu erreichen und aufrechtzuerhalten, und aufgrund der geringen biologischen Wirksamkeit herkömmlicher Aminosäurenmischungen bleibt selbst bei therapietreuen erwachsenen Patienten ein erhöhtes Risiko für Antriebsschwäche, Angstzustände und Aufmerksamkeitsstörungen über die gesamte Lebensspanne bestehen.12
Wie kann die PKU-Behandlung verbessert werden?
Internationale Experten sind sich einig, dass weitere Änderungen erforderlich sind, um die Proteinersatzstoffe zu verbessern und einen entscheidenden Beitrag für Menschen zu leisten, die von PKU betroffen sind.
Das gemeinsame Ziel sollte darin bestehen, optimierte geschmacks- und geruchsneutrale Produkte mit den Eigenschaften natürlicher Proteine anzubieten, die eine stabile chemische Umgebung schaffen, welche mit einer besseren physiologischen Funktion und Verträglichkeit für den Patienten einhergeht7.
Dies bedeutet, dass wir PKU-Produkte anbieten müssen, die den unerfüllten Bedarf an Therapietreue und Stoffwechselkontrolle decken.
Um dieses Ziel zu erreichen und die Lebensqualität der Patienten zu verbessern, hat APR Applied Pharma Research eine pharmazeutische Technologie entwickelt, die zum ersten Mal für ein Lebensmittel für besondere medizinische Zwecke zur Ernährungsbehandlung von PKU eingesetzt wurde.
 Referenzen:
Referenzen:
- Van Wegberg AMJ, MacDonald A, Ahring K, et al. The complete European guidelines on phenylketonuria: diagnosis and treatment. Orphanet J Rare Dis 2017;12(1):162
- Blau N, Hennermann JB, Langenbeck U, Lichter-Konecki U. Diagnosis, classification, and genetics of phenylketonuria and
tetrahydrobiopterin (BH4) deficiencies. Mol Genet Metab. 2011;104
- Walter, John Η., and Fiona J. White. "Blood phenylalanine control in adolescents with phenylketonuria." International journal of adolescent medicine and health 16.1 (2004): 41-46.
- Enns,Greg et al. Molecular genetics and metabolism 101 2-3 (2010): 99-109
- Altman G, Hussain K, Green D, Strauss BJG, Wilcox G. Mental health diagnoses in adults with phenylketonuria: a retrospective systematic audit in a large UK single centre. Orphanet J Rare Dis. 2021 Dec 20;16(1):520
- MacDonald, A., van Wegberg, A.M.J., Ahring, K. et al. PKU dietary handbook to accompany PKU guidelines. Orphanet J Rare
Dis 15, 171 (2020).
- Daly A, et al. Protein substitutes in PKU; their historical evolution. Nutrients. 2021;13(2):484.
- Klimek A, Baerwald C, Schwarz M, et al. Everyday life, dietary practices, and health conditions of adult PKU patients:
a multicenter, cross-sectional study. Ann Nutr Metab 2020;76(4):251–258
- Wu G. Amino acids: metabolism functions, and nutrition. Amino Acids 2009;37(1):1–17
- MacDonald A, Singh RH, Rocha JC, van Spronsen FJ. Optimising amino acid absorption: essential to improve nitrogen balance and metabolic control in phenylketonuria. Nutr Res Rev 2019;32(1):70–78
- Gropper SS and Acosta PB. Effect of simultaneous ingestion of L-amino acids and whole protein on plasma amino acid and urea nitrogen concentrations in humans. J Parenter Enteral Nutr. 1991;15(1):48–53
- Psychiatric and Cognitive Aspects of Phenylketonuria: The Limitations of Diet and Promise of New Treatments. Front Psychiatry. 2019;10:561.
Die tägliche Einhaltung der Diät kann das soziale Leben von PKU-Patienten bei den Mahlzeiten beeinträchtigen, und die regelmäßige Einnahme von Proteinersatzstoffen kann aufgrund ihrer Darreichungsform und Eigenschaften sehr schwierig sein.
Darüber hinaus hat eine kürzlich durchgeführte Studie an insgesamt therapietreuen Patienten gezeigt, dass die Häufigkeit der Einnahme von Aminosäuremischungen bei der PKU-Behandlung eine heikle Rolle spielen kann, da 42 % der erwachsenen PKU-Patienten ihren gesamten Proteinersatz auf ≤2 Dosen/Tag aufteilen (im Gegensatz zu den in den europäischen Leitlinien empfohlenen 3 bis 4 Dosen).8
Ein weiterer wichtiger Aspekt, der bei der PKU-Behandlung zu berücksichtigen ist, betrifft die Art und Weise, wie Aminosäuren nach ihrer Aufnahme über Proteinersatzstoffe vom Körper absorbiert werden.
Personen ohne PKU nehmen natürliche Proteine mit der Nahrung auf (z. B. Fleisch, Fisch, Milchprodukte, Eier usw.), die im Laufe der Zeit verdaut und absorbiert werden (grüner Kasten), so dass der Aminosäurespiegel im Blut bis zu 150 Minuten (2,5 Stunden) konstant bleibt.
Bei PKU-Betroffenen müssen eiweißhaltige Lebensmittel durch freie Aminosäuren ersetzt werden, die in herkömmlichen Proteinersatzprodukten enthalten sind. Proteine und Aminosäuren verhalten sich jedoch sehr unterschiedlich!
Freie Aminosäuren sind wie bereits verdaute Proteine, so dass sie direkt für die Aufnahme durch den Darm verfügbar sind. Dies bedeutet, dass der Aminosäurespiegel im Blut unmittelbar nach dem Verzehr von Proteinersatzprodukten hoch ist9 und innerhalb von nur 30 Minuten einen Höchstwert erreicht, der bald darauf drastisch abfällt (blauer Kasten).1,10
Die Grafik zeigt detaillierter den Unterschied in der Absorptionskinetik zwischen natürlichem Protein (grüne Linie) und freien Aminosäuren (gestrichelte Linie), wobei deren hohe Ausscheidung kurz nach der Aufnahme (Oxidation, roter Bereich) und in den so genannten „Fastenperioden“, angezeigt wird, d. h. in den Zeitabschnitten, in denen die Menge an Aminosäuren im Körper sehr gering ist.10,11
Die Oxidation und schnelle Ausscheidung von Aminosäuren verhindert, dass der Körper sie effizient für physiologische Prozesse wie den Aufbau neuer Proteine nutzen kann, was sich auf den Stoffwechsel des Körpers auswirkt und folglich die Gesundheit beeinträchtigt.10,11
Die unzureichende Absorption von Aminosäuren und die Unfähigkeit des Körpers, sie effizient zu nutzen, ist der Grund, warum in den europäischen Leitlinien für PKU empfohlen wird, herkömmliche Proteinersatzstoffe in kleinen und häufigen Dosen mindestens 3- bis 4-mal täglich zu verzehren.
Die Gesamtproteinzufuhr sollte 40 % mehr als die von FAO/WHO/UNU festgelegten sicheren Eiweißmengen liefern1,6, um den Aminosäurespiegel im Blut konstant zu halten.
Dies ist jedoch oft sehr schwer zu erreichen und aufrechtzuerhalten, und aufgrund der geringen biologischen Wirksamkeit herkömmlicher Aminosäurenmischungen bleibt selbst bei therapietreuen erwachsenen Patienten ein erhöhtes Risiko für Antriebsschwäche, Angstzustände und Aufmerksamkeitsstörungen über die gesamte Lebensspanne bestehen.12
Dies ist jedoch oft sehr schwer zu erreichen und aufrechtzuerhalten, und aufgrund der geringen biologischen Wirksamkeit herkömmlicher Aminosäurenmischungen bleibt selbst bei therapietreuen erwachsenen Patienten ein erhöhtes Risiko für Antriebsschwäche, Angstzustände und Aufmerksamkeitsstörungen über die gesamte Lebensspanne bestehen.12
Wie kann die PKU-Behandlung verbessert werden?
Internationale Experten sind sich einig, dass weitere Änderungen erforderlich sind, um die Proteinersatzstoffe zu verbessern und einen entscheidenden Beitrag für Menschen zu leisten, die von PKU betroffen sind.
Das gemeinsame Ziel sollte darin bestehen, optimierte geschmacks- und geruchsneutrale Produkte mit den Eigenschaften natürlicher Proteine anzubieten, die eine stabile chemische Umgebung schaffen, welche mit einer besseren physiologischen Funktion und Verträglichkeit für den Patienten einhergeht7.
Dies bedeutet, dass wir PKU-Produkte anbieten müssen, die den unerfüllten Bedarf an Therapietreue und Stoffwechselkontrolle decken.
Um dieses Ziel zu erreichen und die Lebensqualität der Patienten zu verbessern, hat APR Applied Pharma Research eine pharmazeutische Technologie entwickelt, die zum ersten Mal für ein Lebensmittel für besondere medizinische Zwecke zur Ernährungsbehandlung von PKU eingesetzt wurde.
Referenzen:
- Van Wegberg AMJ, MacDonald A, Ahring K, et al. The complete European guidelines on phenylketonuria: diagnosis and treatment. Orphanet J Rare Dis 2017;12(1):162
- Blau N, Hennermann JB, Langenbeck U, Lichter-Konecki U. Diagnosis, classification, and genetics of phenylketonuria and tetrahydrobiopterin (BH4) deficiencies. Mol Genet Metab. 2011;104
- Walter, John Η., and Fiona J. White. "Blood phenylalanine control in adolescents with phenylketonuria." International journal of adolescent medicine and health 16.1 (2004): 41-46.
- Enns,Greg et al. Molecular genetics and metabolism 101 2-3 (2010): 99-109
- Altman G, Hussain K, Green D, Strauss BJG, Wilcox G. Mental health diagnoses in adults with phenylketonuria: a retrospective systematic audit in a large UK single centre. Orphanet J Rare Dis. 2021 Dec 20;16(1):520
- MacDonald, A., van Wegberg, A.M.J., Ahring, K. et al. PKU dietary handbook to accompany PKU guidelines. Orphanet J Rare Dis 15, 171 (2020).
- Daly A, et al. Protein substitutes in PKU; their historical evolution. Nutrients. 2021;13(2):484.
- Klimek A, Baerwald C, Schwarz M, et al. Everyday life, dietary practices, and health conditions of adult PKU patients: a multicenter, cross-sectional study. Ann Nutr Metab 2020;76(4):251–258
- Wu G. Amino acids: metabolism functions, and nutrition. Amino Acids 2009;37(1):1–17
- MacDonald A, Singh RH, Rocha JC, van Spronsen FJ. Optimising amino acid absorption: essential to improve nitrogen balance and metabolic control in phenylketonuria. Nutr Res Rev 2019;32(1):70–78
- Gropper SS and Acosta PB. Effect of simultaneous ingestion of L-amino acids and whole protein on plasma amino acid and urea nitrogen concentrations in humans. J Parenter Enteral Nutr. 1991;15(1):48–53
- Psychiatric and Cognitive Aspects of Phenylketonuria: The Limitations of Diet and Promise of New Treatments. Front Psychiatry. 2019;10:561.