

Cos’è la Fenilchetonuria (PKU)?
Sintomi e diagnosi
La fenilchetonuria, detta anche in breve “PKU”, è un disturbo dovuto a un difetto congenito autosomico recessivo nel metabolismo della fenilalanina (Phe). La prevalenza media è di circa 1 paziente ogni 15.000 neonati.
Chi nasce con la Fenilchetonuria non è in grado di scomporre un amminoacido chiamato "fenilalanina” (Phe), di conseguenza quest’ultimo si accumula all’interno dell’organismo in quantità tossiche e provoca gravi danni cerebrali.
Il trattamento si basa prevalentemente su un rigoroso regime alimentare a basso contenuto di proteine (e quindi di Phe), che i pazienti devono seguire per tutta la vita, e una supplementazione proteica (tramite miscele amminoacidiche prive di fenilalanina), necessaria per garantire il normale processo di crescita ed un buono stato di salute.
Una diagnosi precoce e l’adozione immediata delle misure necessarie possono consentire alle persone con PKU di crescere sane e di vivere una vita normale e al massimo delle loro potenzialità.1
Cause e classificazione della Fenilchetonuria
La fenilchetonuria è provocata da variazioni del gene che codifica per la fenilalanina idrossilasi (PAH).
La PAH è un enzima che ha bisogno del cofattore chiamato “tetraidrobiopterina” (BH4) per convertire la fenilalanina (Phe) in tirosina (Tyr). Una carenza o un malfunzionamento di PAH o del suo cofattore BH4 determina un aumento della concentrazione di Phe nel sangue e il raggiungimento di concentrazioni tossiche nel cervello.
Se non viene tenuto attentamente sotto controllo, il livello di Phe nel sangue può diventare significativamente alto, fino a causare problemi neurologici e comportamentali evidenti.1
La PAH è un enzima che ha bisogno del cofattore chiamato “tetraidrobiopterina” (BH4) per convertire la fenilalanina (Phe) in tirosina (Tyr). Una carenza o un malfunzionamento di PAH o del suo cofattore BH4 determina un aumento della concentrazione di Phe nel sangue e il raggiungimento di concentrazioni tossiche nel cervello.
Se non viene tenuto attentamente sotto controllo, il livello di Phe nel sangue può diventare significativamente alto, fino a causare problemi neurologici e comportamentali evidenti.1
A seconda del difetto che interessa l’enzima e della gravità della malattia, sono state individuate diverse forme di PKU con fenotipi clinici diversi. La PKU si suddivide in PKU classica e in forme di PKU più leggere (ad es. la PKU moderata e la PKU lieve).
La definizione del fenotipo di PKU può essere fondamentale per stabilire le opzioni di trattamento, anche in gravidanza.2
Screening neonatale: come si diagnostica la fenilchetonuria?
L’esame per la PKU è generalmente compreso nei test di screening neonatale (SN) che vengono eseguiti in ospedale, tra le 24 e le 72 ore dopo la nascita; si effettua prelevando un campione di sangue dal tallone e misurando i livelli di Phe con una semplice analisi di laboratorio. I programmi di screening neonatale possono variare da Paese a Paese.
Quali sono i sintomi della PKU?
Alla nascita i pazienti PKU non presentano alcun sintomo. Tuttavia, se non vengono sottoposti alle terapie necessarie, lo sviluppo del cervello subisce gravi conseguenze che possono portare a disabilità intellettive e disturbi comportamentali significativi.1
La terapia, soprattutto se seguita nell’arco dell’intero sviluppo del sistema nervoso e anche una volta raggiunta l’età adulta, consente ai pazienti di ottenere buoni risultati a livello cognitivo e psichico.3
Tuttavia, può essere difficile attenersi alla dieta PKU per tutta la vita: infatti, mentre durante l’infanzia viene seguita spesso molto scrupolosamente, la situazione peggiora durante la crescita4. Questo è probabilmente dovuto alla maggiore indipendenza dei pazienti dalle loro famiglie e dal peso della terapia a livello psicologico e sociale.
Tuttavia, può essere difficile attenersi alla dieta PKU per tutta la vita: infatti, mentre durante l’infanzia viene seguita spesso molto scrupolosamente, la situazione peggiora durante la crescita4. Questo è probabilmente dovuto alla maggiore indipendenza dei pazienti dalle loro famiglie e dal peso della terapia a livello psicologico e sociale.
Molti pazienti adolescenti o adulti che interrompono il trattamento o lo seguono in modo meno scrupoloso possono avere sintomi come ansia, depressione e disturbi cognitivi.5
Si può prevenire la PKU?
La PKU è una malattia genetica e non è possibile prevenirla o evitarla. È una condizione ereditaria che si trasmette dai genitori ai figli in modo “recessivo”: per sviluppare la malattia è necessario che entrambi i genitori abbiano una variante mutata del gene della PAH. Le persone con questa caratteristica si chiamano “portatori sani”.
Se solo un genitore è portatore del gene mutato, il figlio non avrà la PKU.
Ma se entrambi i genitori sono portatori sani, c’è una probabilità del 25% che trasmettano al figlio il gene mutato, facendo sì che abbia la PKU.
Ma se entrambi i genitori sono portatori sani, c’è una probabilità del 25% che trasmettano al figlio il gene mutato, facendo sì che abbia la PKU.
PKU e gravidanza
Quando si ha la PKU, la gravidanza va pianificata bene. Le donne con la PKU possono avere bambini sani, purché seguano rigidamente la dieta a basso contenuto di Phe durante il periodo di gestazione.
Se la PKU non viene tenuta sotto controllo adeguatamente durante la gravidanza e se i livelli di fenilalanina nel sangue sono elevati, il feto può subire gravi danni e avere difetti alla nascita.
Se la PKU non viene tenuta sotto controllo adeguatamente durante la gravidanza e se i livelli di fenilalanina nel sangue sono elevati, il feto può subire gravi danni e avere difetti alla nascita.
Più i livelli di Phe nel plasma della madre sono vicini a valori normali, minore sarà la probabilità che si verifichino queste complicanze; per questo motivo, si consiglia di provare a concepire solo quando i livelli di Phe rientrano nell’intervallo corretto. Quando questo non è possibile, ad esempio in caso di gravidanze inattese o non programmate, occorre rivolgersi immediatamente a un centro che si occupa di malattie metaboliche, soprattutto se i livelli di Phe non sono sotto controllo.
In ogni caso, occorre monitorare attentamente la gravidanza, anche se i valori di fenilalanina sono sotto controllo fin dall’inizio: si consiglia una visita ogni trimestre, ma molti medici raccomandano visite più frequenti e l’entità del monitoraggio dipenderà anche dalle esigenze e dal controllo metabolico individuali.1
Il trattamento della PKU
La Phe è un amminoacido, ovvero uno dei mattoni di base che compongono le proteine, ed è presente in tutte le proteine e nella maggior parte degli alimenti; il trattamento prevede quindi una dieta povera di Fenilalanina e particolarmente limitata, che deve essere seguita per tutta la vita con l’obiettivo di mantenere bassi i livelli di Phe nel sangue1,6. La dieta per la PKU prevede:

Fondamentalmente, carne, pesce, uova e latticini non sono consentiti
Pasta, pane e biscotti devono essere “speciali”, ovvero a basso contenuto di proteine

La maggior parte della verdura e della frutta deve essere pesata
La principale fonte di proteine sono i sostituti proteici, generalmente a base di miscele di amminoacidi prive di fenilalanina, che sono essenziali per favorire una normale crescita, evitare carenze proteiche, fornire all’organismo la tirosina e contribuire a tenere sotto controllo in modo ottimale la fenilalanina nel sangue.6

Esigenze non soddisfatte dai sostituti proteici tradizionali
I sostituti proteici convenzionali contengono miscele di amminoacidi liberi e devono essere assunti seguendo le linee guida europee, ovvero in piccole dosi e di frequente, almeno 3-4 volte, distribuite in modo uniforme nell’arco della giornata.1
Oggigiorno esistono molte opzioni disponibili, in diversi gusti e formati; tuttavia, gli esperti internazionali di PKU ritengono che ci sia ancora spazio per un miglioramento, al fine di supportare i pazienti con PKU e rispondere ad alcune esigenze ancora oggi insoddisfatte, relative all’aderenza al trattamento e al controllo metabolico.7
Oggigiorno esistono molte opzioni disponibili, in diversi gusti e formati; tuttavia, gli esperti internazionali di PKU ritengono che ci sia ancora spazio per un miglioramento, al fine di supportare i pazienti con PKU e rispondere ad alcune esigenze ancora oggi insoddisfatte, relative all’aderenza al trattamento e al controllo metabolico.7
1. Aderenza al trattamento
Crescere con la fenilchetonuria può essere difficile, e può diventare un peso invisibile per le persone che convivono con essa.
Seguire quotidianamente una dieta così limitata può influenzare la vita sociale che ruota attorno ai pasti e prendere regolarmente sostituti proteici può essere davvero impegnativo a causa dei formati e delle caratteristiche di questi prodotti.
L’aderenza al trattamento viene influenzata anche dalle caratteristiche organolettiche poco appetibili degli amminoacidi (gusto e odore sgradevoli e retrogusto che persiste a lungo in bocca dopo il consumo)7, che hanno conseguenze negative sulla vita sociale dei PKUers.
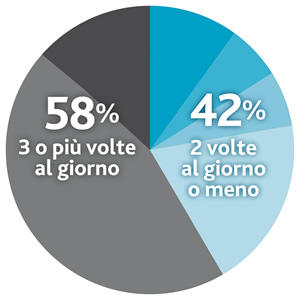 Inoltre, un recente studio condotto su pazienti che seguivano la dieta ha evidenziato che la frequenza del consumo delle miscele di amminoacidi può rivestire un ruolo complesso nel trattamento della PKU: è infatti emerso che il 42% dei pazienti prende la dose completa di sostituti proteici in una o due dosi quotidiane (invece delle 3-4 suggerite dalle linee guida europee).8
Inoltre, un recente studio condotto su pazienti che seguivano la dieta ha evidenziato che la frequenza del consumo delle miscele di amminoacidi può rivestire un ruolo complesso nel trattamento della PKU: è infatti emerso che il 42% dei pazienti prende la dose completa di sostituti proteici in una o due dosi quotidiane (invece delle 3-4 suggerite dalle linee guida europee).8
2. Controllo metabolico
Un altro aspetto importante da considerare, quando si parla del trattamento della PKU, è legato al modo in cui gli amminoacidi vengono assorbiti dall’organismo, dopo essere stati assunti sotto forma di sostituti proteici.
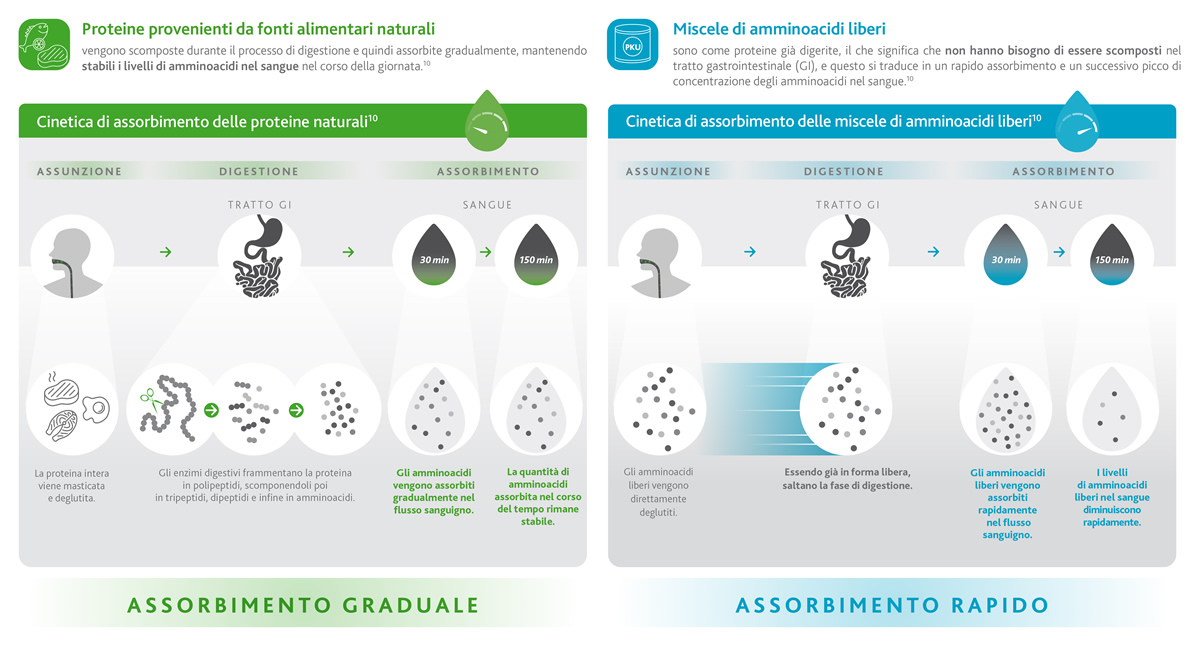
Chi non ha la PKU consuma le proteine naturali presenti negli alimenti (come carne, pesce, latticini, uova, ecc.), che vengono digerite e assorbite gradualmente in un certo arco di tempo (riquadro verde), mantenendo così costanti i livelli di amminoacidi nel sangue per un tempo che può arrivare a 150 minuti (2,5 ore).
Chi ha la PKU deve sostituire gli alimenti a base di proteine con gli amminoacidi liberi contenuti nei sostituti proteici tradizionali. Le proteine e gli amminoacidi liberi si comportano però in modo molto diverso.
Gli amminoacidi liberi sono sostanzialmente proteine già digerite, quindi sono subito disponibili per l’assorbimento nell’intestino. Questo significa che il livello di amminoacidi nel sangue aumenta rapidamente subito dopo aver consumato i sostituti proteici,9 e raggiunge il picco massimo nel giro di soli 30 minuti, per poi diminuire drasticamente poco dopo (riquadro blu).1,10
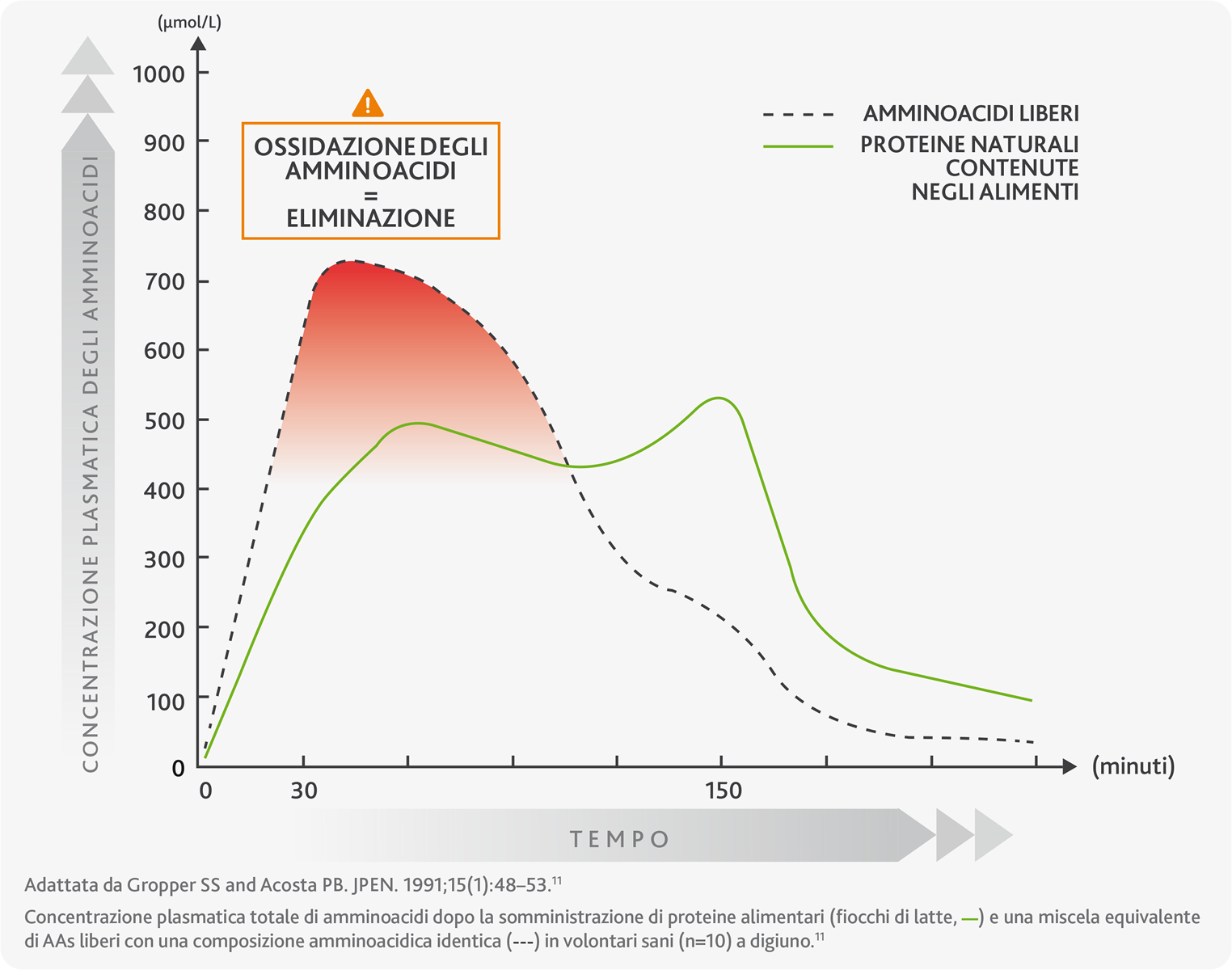
 Il grafico mostra più nel dettaglio come la cinetica dell’assorbimento sia diversa per le proteine naturali (linea verde) e gli amminoacidi liberi (linea tratteggiata), mettendo in risalto l’elevata percentuale di spreco ed eliminazione di questi ultimi (ossidazione, zona rossa) poco dopo l’assunzione e nei cosiddetti 'periodi di digiuno’, periodi di tempo durante i quali la concentrazione di amminoacidi in circolo è molto bassa.10,11
L’ossidazione e la rapida eliminazione degli amminoacidi impediscono all’organismo di utilizzarli in modo efficiente per i processi fisiologici, come la costruzione di nuove proteine. Ne consegue dunque un impatto sul metabolismo e di conseguenza sullo stato di salute.10,11
Il grafico mostra più nel dettaglio come la cinetica dell’assorbimento sia diversa per le proteine naturali (linea verde) e gli amminoacidi liberi (linea tratteggiata), mettendo in risalto l’elevata percentuale di spreco ed eliminazione di questi ultimi (ossidazione, zona rossa) poco dopo l’assunzione e nei cosiddetti 'periodi di digiuno’, periodi di tempo durante i quali la concentrazione di amminoacidi in circolo è molto bassa.10,11
L’ossidazione e la rapida eliminazione degli amminoacidi impediscono all’organismo di utilizzarli in modo efficiente per i processi fisiologici, come la costruzione di nuove proteine. Ne consegue dunque un impatto sul metabolismo e di conseguenza sullo stato di salute.10,11
 L’assorbimento inefficace degli amminoacidi e l’incapacità dell’organismo di utilizzarli in modo efficiente sono i motivi alla base dei suggerimenti presenti nelle linee guida europee, che consigliano di consumare i sostituti proteici tradizionali in piccole dosi e di frequente (3-4 volte al giorno), con lo scopo di mantenere i valori di amminoacidi nel sangue costanti.
In sostanza l’apporto totale di proteine dovrebbe superare del 40% i livelli di proteine ritenuti sicuri da FAO/OMS/UNU1,6
L’assorbimento inefficace degli amminoacidi e l’incapacità dell’organismo di utilizzarli in modo efficiente sono i motivi alla base dei suggerimenti presenti nelle linee guida europee, che consigliano di consumare i sostituti proteici tradizionali in piccole dosi e di frequente (3-4 volte al giorno), con lo scopo di mantenere i valori di amminoacidi nel sangue costanti.
In sostanza l’apporto totale di proteine dovrebbe superare del 40% i livelli di proteine ritenuti sicuri da FAO/OMS/UNU1,6
Tuttavia, questo è un obiettivo spesso molto difficile da raggiungere e mantenere. Talvolta, a causa della scarsa efficacia biologica delle miscele di amminoacidi tradizionali, persino per i pazienti adulti che seguono con scrupolo la dieta, continua a esistere un rischio elevato di depressione, ansia e disturbi dell’attenzione.12
Come si può migliorare la terapia per la PKU?
A livello internazionale, gli esperti sono concordi sul fatto che sia necessario migliorare i sostituti proteici affinché possano fare davvero la differenza nelle vite delle persone che convivono con la PKU e che hanno quotidianamente a che fare con essa.
L’obiettivo comune a cui puntare è offrire prodotti inodori e con un gusto ottimizzato, che abbiano le proprietà delle proteine naturali per creare un ambiente chimico stabile associato ad una migliore funzionalità fisiologica e aumentare la tolleranza da parte del paziente7.
Questo vuol dire offrire prodotti in grado di rispondere alle esigenze ancora insoddisfatte sul piano dell’aderenza al trattamento e del controllo metabolico.
Per raggiungere questo obiettivo e migliorare la qualità della vita dei pazienti, APR (Applied Pharma Research) ha sviluppato una tecnologia farmaceutica, che per la prima volta è stata applicata ad un alimento a fini medici speciali indicato per il trattamento della PKU.
 Bibliografia:
Bibliografia:
- Van Wegberg AMJ, MacDonald A, Ahring K, et al. The complete European guidelines on phenylketonuria: diagnosis and treatment. Orphanet J Rare Dis 2017;12(1):162
- Blau N, Hennermann JB, Langenbeck U, Lichter-Konecki U. Diagnosis, classification, and genetics of phenylketonuria and
tetrahydrobiopterin (BH4) deficiencies. Mol Genet Metab. 2011;104
- Walter, John Η., and Fiona J. White. "Blood phenylalanine control in adolescents with phenylketonuria." International journal of adolescent medicine and health 16.1 (2004): 41-46.
- Enns,Greg et al. Molecular genetics and metabolism 101 2-3 (2010): 99-109
- Altman G, Hussain K, Green D, Strauss BJG, Wilcox G. Mental health diagnoses in adults with phenylketonuria: a retrospective systematic audit in a large UK single centre. Orphanet J Rare Dis. 2021 Dec 20;16(1):520
- MacDonald, A., van Wegberg, A.M.J., Ahring, K. et al. PKU dietary handbook to accompany PKU guidelines. Orphanet J Rare
Dis 15, 171 (2020).
- Daly A, et al. Protein substitutes in PKU; their historical evolution. Nutrients. 2021;13(2):484.
- Klimek A, Baerwald C, Schwarz M, et al. Everyday life, dietary practices, and health conditions of adult PKU patients:
a multicenter, cross-sectional study. Ann Nutr Metab 2020;76(4):251–258
- Wu G. Amino acids: metabolism functions, and nutrition. Amino Acids 2009;37(1):1–17
- MacDonald A, Singh RH, Rocha JC, van Spronsen FJ. Optimising amino acid absorption: essential to improve nitrogen balance and metabolic control in phenylketonuria. Nutr Res Rev 2019;32(1):70–78
- Gropper SS and Acosta PB. Effect of simultaneous ingestion of L-amino acids and whole protein on plasma amino acid and urea nitrogen concentrations in humans. J Parenter Enteral Nutr. 1991;15(1):48–53
- Psychiatric and Cognitive Aspects of Phenylketonuria: The Limitations of Diet and Promise of New Treatments. Front Psychiatry. 2019;10:561.
Seguire quotidianamente una dieta così limitata può influenzare la vita sociale che ruota attorno ai pasti e prendere regolarmente sostituti proteici può essere davvero impegnativo a causa dei formati e delle caratteristiche di questi prodotti.
Inoltre, un recente studio condotto su pazienti che seguivano la dieta ha evidenziato che la frequenza del consumo delle miscele di amminoacidi può rivestire un ruolo complesso nel trattamento della PKU: è infatti emerso che il 42% dei pazienti prende la dose completa di sostituti proteici in una o due dosi quotidiane (invece delle 3-4 suggerite dalle linee guida europee).8
Un altro aspetto importante da considerare, quando si parla del trattamento della PKU, è legato al modo in cui gli amminoacidi vengono assorbiti dall’organismo, dopo essere stati assunti sotto forma di sostituti proteici.
Chi non ha la PKU consuma le proteine naturali presenti negli alimenti (come carne, pesce, latticini, uova, ecc.), che vengono digerite e assorbite gradualmente in un certo arco di tempo (riquadro verde), mantenendo così costanti i livelli di amminoacidi nel sangue per un tempo che può arrivare a 150 minuti (2,5 ore).
Chi ha la PKU deve sostituire gli alimenti a base di proteine con gli amminoacidi liberi contenuti nei sostituti proteici tradizionali. Le proteine e gli amminoacidi liberi si comportano però in modo molto diverso.
Gli amminoacidi liberi sono sostanzialmente proteine già digerite, quindi sono subito disponibili per l’assorbimento nell’intestino. Questo significa che il livello di amminoacidi nel sangue aumenta rapidamente subito dopo aver consumato i sostituti proteici,9 e raggiunge il picco massimo nel giro di soli 30 minuti, per poi diminuire drasticamente poco dopo (riquadro blu).1,10
Il grafico mostra più nel dettaglio come la cinetica dell’assorbimento sia diversa per le proteine naturali (linea verde) e gli amminoacidi liberi (linea tratteggiata), mettendo in risalto l’elevata percentuale di spreco ed eliminazione di questi ultimi (ossidazione, zona rossa) poco dopo l’assunzione e nei cosiddetti 'periodi di digiuno’, periodi di tempo durante i quali la concentrazione di amminoacidi in circolo è molto bassa.10,11
L’ossidazione e la rapida eliminazione degli amminoacidi impediscono all’organismo di utilizzarli in modo efficiente per i processi fisiologici, come la costruzione di nuove proteine. Ne consegue dunque un impatto sul metabolismo e di conseguenza sullo stato di salute.10,11
L’assorbimento inefficace degli amminoacidi e l’incapacità dell’organismo di utilizzarli in modo efficiente sono i motivi alla base dei suggerimenti presenti nelle linee guida europee, che consigliano di consumare i sostituti proteici tradizionali in piccole dosi e di frequente (3-4 volte al giorno), con lo scopo di mantenere i valori di amminoacidi nel sangue costanti.
In sostanza l’apporto totale di proteine dovrebbe superare del 40% i livelli di proteine ritenuti sicuri da FAO/OMS/UNU1,6
Tuttavia, questo è un obiettivo spesso molto difficile da raggiungere e mantenere. Talvolta, a causa della scarsa efficacia biologica delle miscele di amminoacidi tradizionali, persino per i pazienti adulti che seguono con scrupolo la dieta, continua a esistere un rischio elevato di depressione, ansia e disturbi dell’attenzione.12
Tuttavia, questo è un obiettivo spesso molto difficile da raggiungere e mantenere. Talvolta, a causa della scarsa efficacia biologica delle miscele di amminoacidi tradizionali, persino per i pazienti adulti che seguono con scrupolo la dieta, continua a esistere un rischio elevato di depressione, ansia e disturbi dell’attenzione.12
Come si può migliorare la terapia per la PKU?
A livello internazionale, gli esperti sono concordi sul fatto che sia necessario migliorare i sostituti proteici affinché possano fare davvero la differenza nelle vite delle persone che convivono con la PKU e che hanno quotidianamente a che fare con essa.
L’obiettivo comune a cui puntare è offrire prodotti inodori e con un gusto ottimizzato, che abbiano le proprietà delle proteine naturali per creare un ambiente chimico stabile associato ad una migliore funzionalità fisiologica e aumentare la tolleranza da parte del paziente7.
Questo vuol dire offrire prodotti in grado di rispondere alle esigenze ancora insoddisfatte sul piano dell’aderenza al trattamento e del controllo metabolico.
Per raggiungere questo obiettivo e migliorare la qualità della vita dei pazienti, APR (Applied Pharma Research) ha sviluppato una tecnologia farmaceutica, che per la prima volta è stata applicata ad un alimento a fini medici speciali indicato per il trattamento della PKU.
Bibliografia:
- Van Wegberg AMJ, MacDonald A, Ahring K, et al. The complete European guidelines on phenylketonuria: diagnosis and treatment. Orphanet J Rare Dis 2017;12(1):162
- Blau N, Hennermann JB, Langenbeck U, Lichter-Konecki U. Diagnosis, classification, and genetics of phenylketonuria and tetrahydrobiopterin (BH4) deficiencies. Mol Genet Metab. 2011;104
- Walter, John Η., and Fiona J. White. "Blood phenylalanine control in adolescents with phenylketonuria." International journal of adolescent medicine and health 16.1 (2004): 41-46.
- Enns,Greg et al. Molecular genetics and metabolism 101 2-3 (2010): 99-109
- Altman G, Hussain K, Green D, Strauss BJG, Wilcox G. Mental health diagnoses in adults with phenylketonuria: a retrospective systematic audit in a large UK single centre. Orphanet J Rare Dis. 2021 Dec 20;16(1):520
- MacDonald, A., van Wegberg, A.M.J., Ahring, K. et al. PKU dietary handbook to accompany PKU guidelines. Orphanet J Rare Dis 15, 171 (2020).
- Daly A, et al. Protein substitutes in PKU; their historical evolution. Nutrients. 2021;13(2):484.
- Klimek A, Baerwald C, Schwarz M, et al. Everyday life, dietary practices, and health conditions of adult PKU patients: a multicenter, cross-sectional study. Ann Nutr Metab 2020;76(4):251–258
- Wu G. Amino acids: metabolism functions, and nutrition. Amino Acids 2009;37(1):1–17
- MacDonald A, Singh RH, Rocha JC, van Spronsen FJ. Optimising amino acid absorption: essential to improve nitrogen balance and metabolic control in phenylketonuria. Nutr Res Rev 2019;32(1):70–78
- Gropper SS and Acosta PB. Effect of simultaneous ingestion of L-amino acids and whole protein on plasma amino acid and urea nitrogen concentrations in humans. J Parenter Enteral Nutr. 1991;15(1):48–53
- Psychiatric and Cognitive Aspects of Phenylketonuria: The Limitations of Diet and Promise of New Treatments. Front Psychiatry. 2019;10:561.